Software
In addition to the archived projects below, for cutting-edge, in-development, and recently released work, check out our organization page on GitHub!
![]()
omniClassifier
Robust prediction models are important for numerous science, engineering, and biomedical applications. However, best-practice procedures for optimizing prediction models can be computationally complex, especially when choosing models from among hundreds or thousands of parameter choices. Computational complexity has further increased with the growth of data in these fields, concurrent with the era of “Big Data”. Grid computing is a potential solution to the computational challenges of Big Data. Desktop grid computing, which uses idle CPU cycles of commodity desktop machines, coupled with commercial cloud computing resources can enable research labs to gain easier and more cost effective access to vast computing resources. We have developed omniClassifier, a multi-purpose prediction modeling application that provides researchers with a tool for conducting machine learning research within the guidelines of recommended best-practices. omniClassifier is implemented as a desktop grid computing system using the Berkeley Open Infrastructure for Network Computing (BOINC) middleware.

caCORRECT
For laboratories that produce many microarray chip data files, quality control is central to valid results. Some rely on quality control provided by microarray manufacturers and scanner hardware. Others use statistics software such as dChip, or routines provided in bioconductor (RMA, MAS, PLIER) to detect outliers in these experiments. caCORRECT represents the next-generation of microarray quality control technology that fuses spatial artifact detection (similar to Harshlighting) and model-based techniques to provide improved gene expression data quality in the presence of artifacts.

- Stokes TH, *Moffitt RA (equal contributing authors), Phan JH, and Wang MD. chip artifact CORRECTion (caCORRECT): A bioinformatics system for quality assurance of genomics and proteomics array data. Ann Biomed Eng. 2007 Jun; 35(6):1068-80.
- Moffitt RA, Yin-Goen Q, Stokes TH, Parry RM, Torrance JH, Phan JH, Young AN, and Wang MD. caCORRECT2: improving accuracy and reliability of microarray data in the presence of artifacts. BMC Bioinformatics. 2011 Sep 29; 12:383.
omniBiomarker
OmniBiomarker is a web-based bioinformatics application that addresses the microarray “curse of dimensionality” problem as well as the software standards problem. Biomarker identification from high-throughput microarray data for clinical prediction is sensitive to analysis parameters. As a result, candidate biomarker lists can be difficult to reproduce, limiting the efficiency of translating candidate biomarker lists to clinical applications. OmniBiomarker addresses this problem by tuning steps in the analysis pipeline to a clinical problem based on prior biological knowledge. By integrating knowledge in this manner, we can overcome the .curse-of-dimensionality. problem and increase the reproducibility of biomarker identification and clinical prediction. Furthermore, omniBiomarker will also include functionality for knowledge-driven data combination to increase the statistical power of biomarker identification. Finally, omniBiomarker addresses the problem of community accessibility. It is focused on not only the novelty of the analysis pipeline, but also on the integration of these analytical steps into a user-friendly, web-accessible interface. OmniBiomarker is now caBIG Silver level compliant, further increasing the interoperability of its functions with other bioinformatics tools in the cancer research community.

- Phan JH, Yin-Goen Q, Young AN, and Wang MD. Improving the efficiency of biomarker identification using biological knowledge. Pac Symp Biocomput. 2009:427-38.
- Phan JH, Moffitt RA, Stokes TH, Liu J, Young AN, *Nie SM, and *Wang MD (co-corresponding senior authors). Convergence of biomarkers, bioinformatics and nanotechnology for individualized cancer treatment. Trends Biotechnol. 2009 Jun; 27(6):350-8.
- Phan JH, Young AN, and Wang MD. omniBiomarker: a web-based application for knowledge-driven biomarker identification. IEEE Trans Biomed Eng. In Press.
- Phan JH, Young AN, and Wang MD. Robust microarray meta-analysis identifies differentially expressed genes for clinical prediction. ScientificWorldJournal, Bioinformatics and Biomedical Informatics. 2012 Nov 28; 2012:989637.
SMART-on-FHIR Mortality Reporting Application
Developed in collaboration with the Centers for Disease Control and Prevention (CDC), this application aims to streamline the process and provide decision support at the time of completing a patient’s death certificate.
ArrayWiki and TissueWiki
Wikis are valuable for data management because: 1) they have an intuitive interface for users to browse and contribute information, 2) they are searchable by Google and other web index/search engines and 3) they come with many tools to ensure data source attribution, conflict resolution, and data integrity maintenance. In the near future, if efforts such as the dbPedia project are successful, Wikis may be as useful as relational databases for executing queries on large repositories.
We seed our Wikis with data from public data repositories: ArrayWiki with Gene Expression Omnibus (GEO) and TissueWiki with Human Protein Atlas (HPA). We offer additional meta-data not offered by any other repository. We calculate data quality scores and combine data compression with data visualization in a novel data format based on open standards. The data quality scores allow users to better discriminate between analyses of low or high confidence.

- Stokes TH, Torrance JT, Li H, and Wang MD. ArrayWiki: an enabling technology for sharing public microarray data repositories and meta-analyses. BMC Bioinformatics. 2008 May 28; 9(Suppl 6):S18.
- Stokes TH, Moffitt RA, Hang S, and Wang MD. TissueWiki: a community repository of semantically annotated tissue images. BMC Bioinformatics. Under Review.
omniSpect
Multispectral imaging technologies capture spatial as well as spectral information from a sample. For example, quantum dots target specific biomarkers and emit different fluorescent spectra; or, mass spectrometry reveals different molecular distributions within a sample. OmniSpect untangles the contributions from different sources within a sample by leveraging their known spectral profiles or inferring them directly from the data.

Q-IHC
Imaging modalities have been at the forefront of the fight against cancer by providing physicians with a variety of methods to make cancer diagnosis and prognosis. The increase in technology capabilities and data volume has lead to a critical need to help physicians make full use of the overwhelming information at hand. In addition, the traditional cancer diagnosis methods depend heavily on expert training, suffers from inter-observer variability, subjectivity and procedural inconsistencies. Quantitative cancer image analysis promises to address these issues by bringing consistency and accuracy to the results. Q-IHC is a set of cancer imaging analysis tools to assess Quantum Dots (QD) and Immunohistochemistry (IHC) based molecular and tissue images for various cancers types. This includes tools for semi-automatic segmentation, morphological feature analysis and quantification, color based region detection, automatic cell counting, and quantitative molecular profiling. With special emphasis on usability and usefulness, these tools will help accelerate research and contribute significantly to the fight against cancer.
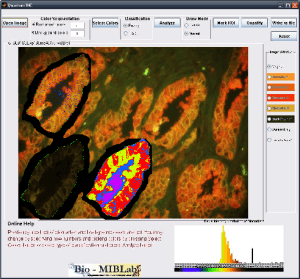
Downloads: Coming Soon
DetectTLC
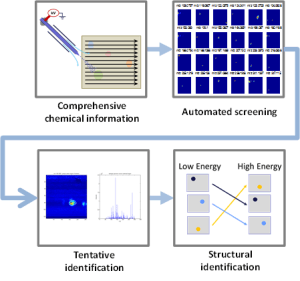
Compared to other types of -omics data, the “chemical fingerprints” measured in metabolomics are highly dynamic, informative, and promising for basic research, disease diagnosis, and disease monitoring. The integration of mass spectrometry imaging (MSI) and thin layer chromatography (TLC) provides a valuable tool for metabolomics research. However, extracting knowledge from MSI-TLC datasets is difficult due to the size (i.e., tens of thousands of images) and the high number of non-informative images. Thus, we present DetectTLC, the first software tool for extracting images from MSI datasets corresponding to TLC spots. This tool allows both completely automated mining and interactive exploration of MSI-TLC datasets. DetectTLC can enable and accelerate research in metabolomics and other applications utilizing MSI-TLC data.
Click here to download DetectTLC.
DataImpute

Quality issues, such as missing information and erroneous data entry, which adversely affect the downstream processing and predictive modeling. Conventional missing data interpolation and imputation techniques perform poorly because there are no standards for modeling the missing data. Current models for imputing missing data include multiple imputation, expectation maximization, and hot – deck imputation techniques. However, they do not account for the type of missing data, which can lead to bias. In our study, we assign the missing data into three categories: “Neglectable” also known in the literature as missing completely at random, “Recoverable” also known as missing at random and “Not Easily Recoverable” also as missing not at random. Then we propose imputation techniques for each type of missing data. We demonstrate our results on a publicly available source of ICU data called MIMIC II using Random Forests for prediction. Our results indicate that our novel imputation techniques outperformed standard mean filling techniques and expectation maximization with a statistical significance p ≤.01 for predicting ICU mortality.