Gene Expression Microarrays
Algorithms, tools, and infrastructure for gene expression microarrays
- caCORRECT: artifact detection and correction in microarrays
- ArrayWiki: community annotation of microarray data
- Knowledge-based biomarker identification
- Meta-analysis of gene expression microarrays
- omniBiomarker: knowledge-based and meta-analysis of gene expression microarrays
- GoMiner: biological interpretation of genomic and proteomic data
caCORRECT: Artifact Detection and Correction in Gene Expression Microarrays
Quality assurance of high throughput -omics data is a major concern for biomedical discovery and translational medicine, and is considered a top priority in bioinformatics and systems biology. We developed a web-based bioinformatics tool called caCORRECT for chip artifact detection, analysis, and CORRECTion, which removes systematic artifactual noise that are commonly observed in microarray gene expression data. caCORRECT improves the reproducibility and reliability of experimental results across several common Affymetrix microarray platforms. caCORRECT represents an advance over state-of-the-art quality control methods such as Harshlighting, and acts to improve gene expression calculation techniques such as PLIER, RMA and MAS5.0, because it incorporates spatial information into outlier detection as well as outlier information into probe normalization. We assessed caCORRECT’s ability to recover accurate gene expression from low-quality probe intensity data using a combination of real and synthetic artifacts with PCR validation and affycomp spike-in data.

http://cacorrect.bme.gatech.edu/
*Stokes TH, *Moffitt RA (equal contributing authors), Phan JH, and Wang MD. “chip artifact CORRECTion (caCORRECT): A bioinformatics system for quality assurance of genomics and proteomics array data.” Ann Biomed Eng. 2007 Jun; 35(6): 1068-80.
ArrayWiki: An Enabling Technology for Sharing Public Microarray Data Repositories and Meta-Analysis
We have utilized Wikis to create ArrayWiki, which enables the bioinformatics community to actively annotate and contribute to the management of the large volumes of microarray data currently available from public repositories. Wikis are valuable for data management because: (1) they have an intuitive interface for users to browse and contribute information, (2) they are searchable by Google and other web index/search engines, and (3) they come with many tools to ensure data source attribution, conflict resolution, and data integrity maintenance. We have seeded ArrayWiki using the Gene Expression Omnibus (GEO) and offer additional meta-data not offered by any other repository. We calculate data quality scores and combine data compression with data visualization in a novel data format based on open standards. The data quality scores allow users to better discriminate between analyses of low or high confidence.

http://arraywiki.bme.gatech.edu/
Improving the Efficiency of Biomarker Identification Using Biological Knowledge
Identifying and validating biomarkers from high-throughput gene expression data is important for understanding and treating cancer. Typically, we identify candidate biomarkers as features that are differentially expressed between two or more classes of samples. Many feature selection metrics rely on ranking by some measure of differential expression. However, interpreting these results is difficult due to the large variety of existing algorithms and metrics, each of which may produce different results. Consequently, a feature ranking metric may work well on some datasets but perform considerably worse on others. We developed a method to choose an optimal feature ranking metric on an individual dataset basis. A metric is optimal if, for a particular dataset, it favorably ranks features that are known to be relevant biomarkers. Extensive knowledge of biomarker candidates is available in public databases and literature. Using this knowledge, we chose a ranking metric that produced the most biologically meaningful results. We first developed a framework for assessing the ability of a ranking metric to detect known relevant biomarkers. We then applied this method to clinical renal cancer microarray data to choose an optimal metric and identified several candidate biomarkers.
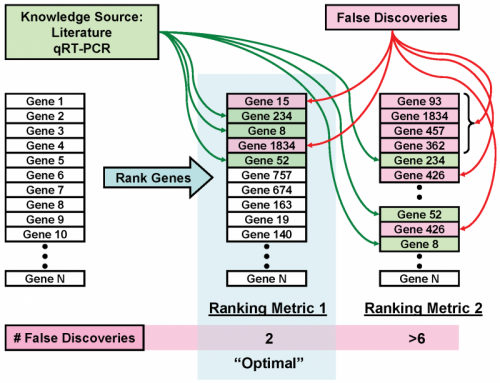
Phan JH, Yin-Goen Q, *Young AN, and *Wang MD (co-corresponding senior authors). “Improving the efficiency of biomarker identification using biological knowledge.” Pac Symp Biocomput, PSB. Kona, HI, USA. 2009 Jan 5; 14: 427-38.
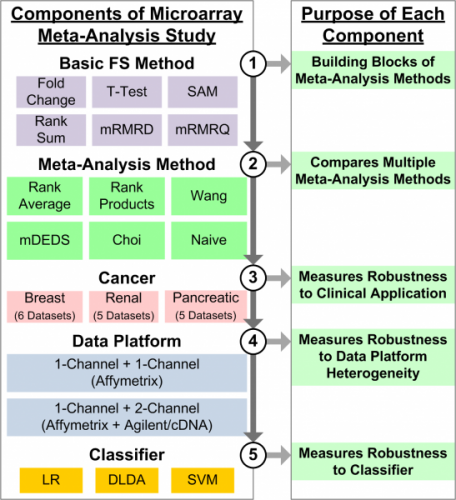
Meta-Analysis of Gene Expression Microarrays
Combining multiple microarray datasets increases sample size and leads to improved reproducibility in identification of informative genes and subsequent clinical prediction. Although microarrays have increased the rate of genomic data collection, sample size is still a major issue when identifying informative genetic biomarkers. Because of this, feature selection methods often suffer from false discoveries, resulting in poorly performing predictive models. We developed a simple meta-analysis-based feature selection method that captures the knowledge in each individual dataset and combines the results using a simple rank average. In a comprehensive study that measures robustness in terms of clinical application (i.e., breast, renal, and pancreatic cancer), microarray platform heterogeneity, and classifier (i.e., logistic regression, diagonal LDA, and linear SVM), we compared the rank average meta-analysis method to five other meta-analysis methods. Results indicated that rank average meta-analysis consistently performed well compared to five other meta-analysis methods.
omniBiomarker: a Web-Based Application for Knowledge-Driven Biomarker Identification
OmniBiomarker is a web-based bioinformatics application that addresses the -omics ”curse of dimensionality“ problem as well as the software standards problem. Biomarker identification from high-throughput gene expression data for clinical prediction is sensitive to analysis parameters. As a result, candidate biomarker lists can be difficult to reproduce, limiting the efficiency of translating candidate biomarker lists to clinical applications. OmniBiomarker addresses this problem by tuning steps in the analysis pipeline to a clinical problem based on prior biological knowledge. By integrating knowledge in this manner, we can overcome the curse-of-dimensionality problem and increase the reproducibility of biomarker identification and clinical prediction. Furthermore, omniBiomarker includes functionality for knowledge-driven data combination to increase the statistical power of biomarker identification. Finally, omniBiomarker addresses the problem of community accessibility by integrating all analytical steps into a user-friendly, web-based interface.
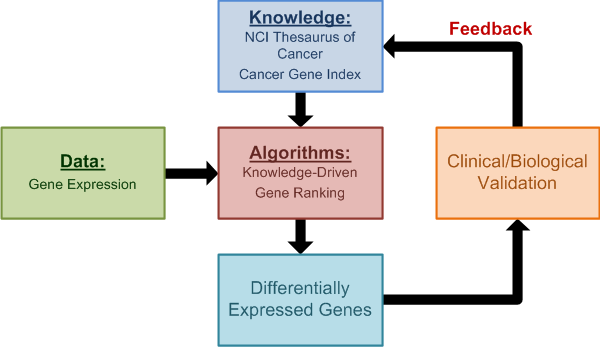

http://omnibiomarker.bme.gatech.edu/
GoMiner: A Resource for Biological Interpretation of Genomic and Proteomic Data
With the growth of genomic data sizes and post-processing results, it is increasingly important to interpret the biological relevance of bioinformatics results. We have developed GoMiner, a program package that organizes lists of ‘interesting’ genes (for example, under- and overexpressed genes from a microarray experiment) for biological interpretation in the context of the Gene Ontology. GoMiner provides quantitative and statistical output files and two useful visualizations. The first is a tree-like structure analogous to that in the AmiGO browser and the second is a compact, dynamically interactive ‘directed acyclic graph’. Genes displayed in GoMiner are linked to major public bioinformatics resources.
http://discover.nci.nih.gov/gominer/index.jsp